
Los términos de este Glosario, disponibles en orden alfabético, se elaboraron a partir de las entradas encontradas en el sitio y en las publicaciones de la Red de Genómica de Fiocruz. El contenido de esta página está en constante revisión y con el tiempo se añadirán nuevos términos.
Amplicón
Nombre dado a los trozos de ADN amplificados en las reacciones de PCR. Tanto los productos de la reacción como las secuencias utilizadas como modelos reciben el nombre de “amplicón”.
Análisis Bayesiano
La estadística bayesiana es un enfoque del tratamiento de datos en el que, a diferencia de la estadística clásica (frecuentista), el conocimiento previo sobre el fenómeno estudiado se tiene en cuenta explícitamente en los análisis. Este enfoque puede proporcionar análisis mejor fundamentados, ya que tiene en cuenta modelos construidos a partir de datos anteriores (por ejemplo, de investigaciones previas) en lugar de las distribuciones hipotéticas utilizadas en las pruebas estadísticas frecuentistas. Desde el punto de vista del análisis filogenético, el análisis bayesiano implica la incorporación de conocimientos previos sobre grupos de organismos (o virus) para obtener conclusiones sobre el parentesco de las nuevas muestras analizadas, tanto entre sí como en relación con los organismos previamente conocidos.
Ancestro común
En biología evolutiva, un “ancestro común” es aquel del que derivan otros organismos. Esta ascendencia puede darse a diferentes escalas, por lo que se puede hablar de:
- Grupos enteros de organismos que derivan de ancestros comunes (como en el caso de las aves, que descienden de los dinosaurios);
- Especies del mismo grupo que comparten ancestros comunes. Por ejemplo, tres de las especies del género Panthera, los leones, los leopardos y los jaguares, descienden de un mismo ancestro, que es diferente de las especies que dieron lugar a los tigres y a los leopardos de las nieves. A su vez, estas dos especies ancestrales también derivan del mismo ancestro común;
- Linajes dentro de la misma especie, como en el caso de los linajes del SARS-CoV-2, causantes del COVID-19. A partir de un genoma viral común, se derivaron nuevos linajes por la acumulación de mutaciones genéticas, de modo que fue posible agrupar las variantes en conjuntos según sus historias genéticas comunes;
Bioinformática
La bioinformática es el nombre que reciben las diversas aplicaciones informáticas desarrolladas para el estudio de la biología. Los usos de la bioinformática van desde programas informáticos para el análisis de secuencias genómicas y su comparación con secuencias similares ya identificadas hasta la modelización tridimensional de la estructura de biomoléculas como las proteínas y los azúcares. En el contexto de la Red Genómica, la bioinformática se utiliza principalmente para analizar y comparar los genomas de diferentes muestras del nuevo coronavirus SARS-CoV-2, con el fin de identificar las mutaciones características de las nuevas variantes e incluso detectar nuevas mutaciones. El uso de herramientas de modelado tridimensional de proteínas también está ganando más espacio en la Red de Genómica, lo que permitirá estudiar en profundidad la proteína Spike del nuevo coronavirus -que permite su entrada en las células- y los efectos de las mutaciones en la estructura y funcionalidad de esta proteína, con posible aumento o disminución de la infectividad.
Coronavirus
Grupo de virus (subfamilia Orthocoronaviridae) descubierto en 1937, asociado principalmente a enfermedades respiratorias en animales, incluida la especie humana. La pandemia actual está causada por el virus denominado SARS-CoV-2, debido a su similitud con el agente que causó la epidemia de SARS (Síndrome Respiratorio Agudo Severo) en 2003.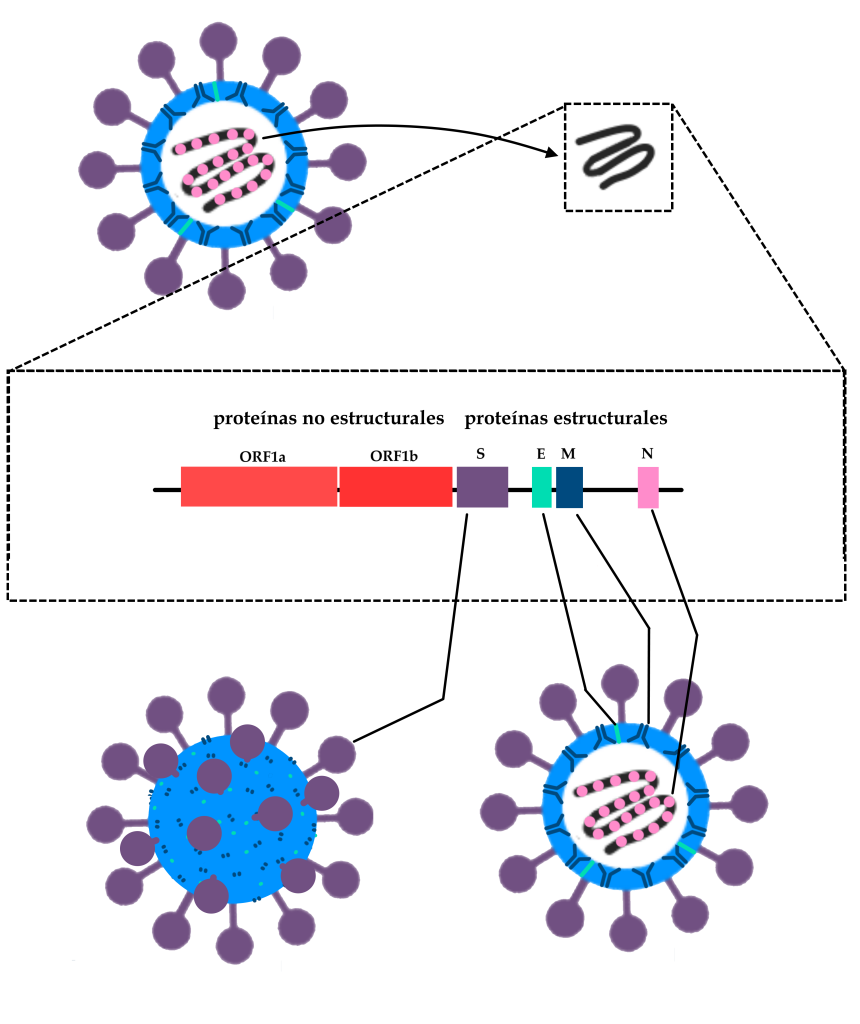
Esquema simplificado de la estructura genómica del SARS-CoV-2, destacando los genes que codifican la glicoproteína de la Spike (S), la proteína de la envoltura (E), la proteína de la membrana (M) y la proteína que envuelve el RNA dentro de la partícula viral completa, formando la nucleocápside (N). También se destacan las regiones del genoma denominadas ORF1a y ORF1b, donde se encuentran parte de los genes de las proteínas no estructurales del virus, es decir, las que no forman parte de la estructura de la partícula viral. Estas proteínas tienen diferentes funciones en la infección, como hacer copias del ARN viral dentro de la célula y redirigir el metabolismo celular para convertir la célula en una “fábrica de virus”.
Antes del SARS-CoV-2, se identificaron otros seis coronavirus capaces de causar infecciones en humanos, cuatro de los cuales estaban implicados en afecciones leves similares al resfriado común. Además de estos cuatro coronavirus con perfiles de enfermedad más leves y del SARS-CoV (o SARS-CoV-1), causante de la epidemia de 2003 caracterizada por un cuadro respiratorio más grave, el otro coronavirus causante de la enfermedad en humanos es el agente causante del MERS (Síndrome Respiratorio de Oriente Medio), una epidemia asociada a la cría de camellos y con tasas de mortalidad más altas (37%) que el SARS y el COVID-19.
El coronavirus entra en las células a partir de la unión de la proteína llamada Spike (o S), con un dominio que se une a la enzima convertidora de angiotensina 2 -una proteína que se encuentra en la superficie de las células humanas y que actúa como “receptor” del virus- y otro dominio que hace que se produzca la fusión entre la membrana celular y la partícula viral, permitiendo la entrada del material genético (RNA) del virus y el inicio de la infección.
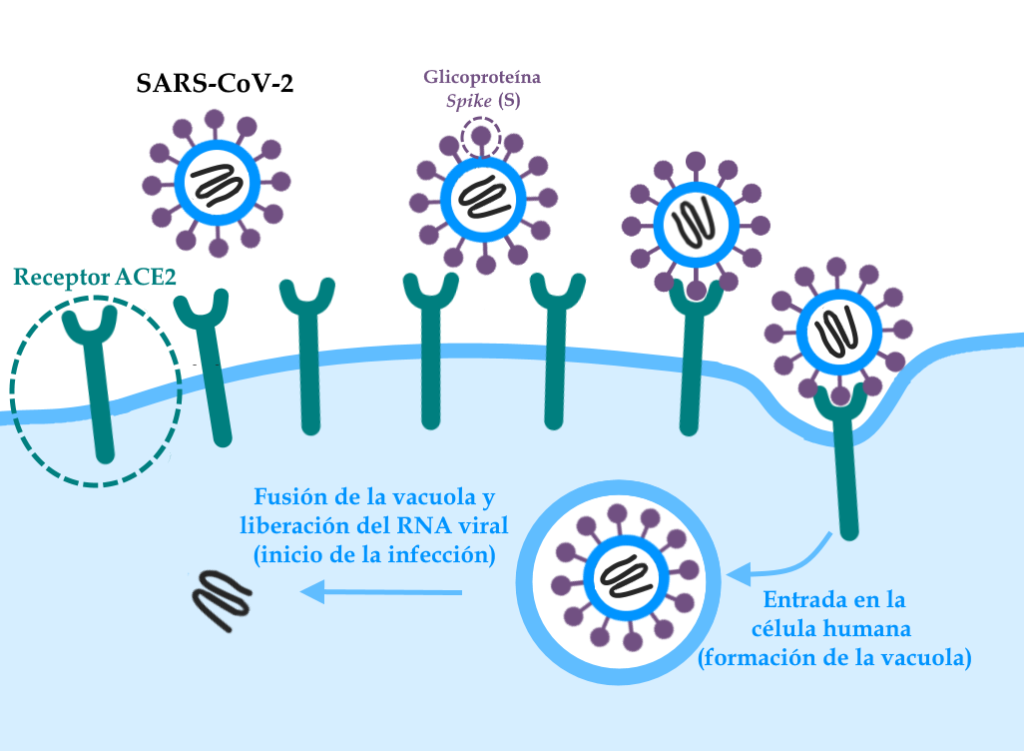
Esquema que ilustra la unión entre la glicoproteína Spike (proteína S) del SARS-CoV-2 y la enzima convertidora de angiotensina 2 (ACE-2) presente en las células humanas. Tras la unión, la célula internaliza la partícula viral en una vacuola, que luego se fusiona con la envoltura viral, liberando el material genético que iniciará la infección.
La hiperinflamación característica de la COVID-19 es uno de los principales mecanismos patogénicos de la enfermedad, lo que provoca fiebre y pérdida de la función respiratoria, que a su vez puede llevar a la necesidad de hospitalización y ventilación mecánica en los casos más graves. La producción excesiva de citoquinas -moléculas de comunicación entre las células, implicadas en la regulación de los mecanismos inmunitarios- asociada a los estados inflamatorios está directamente relacionada con la fiebre y la afectación de los tejidos seguida de fibrosis, en un fenómeno denominado “tormenta de citoquinas”. La tormenta de citoquinas se asocia a otras afecciones virales que tienen efectos hiperinflamatorios como parte importante de la patogénesis, como los casos graves de dengue, por ejemplo.
Se cree que el SARS-CoV-2 se originó en la especie de los murciélagos y, debido a las mutaciones, fue capaz de infectar y transmitirse entre otras especies de mamíferos intermedios, incluyendo finalmente la especie humana. Los análisis de las muestras que circulan entre otras poblaciones animales sitúan al pangolín malayo como posible intermediario en este proceso de adaptación a otros huéspedes (también llamado spillover, del inglés para “rebosar”). La pandemia de COVID-19, con más de 2 millones de muertes confirmadas en todo el mundo hasta enero de 2021, es la mayor experimentada por la humanidad desde la primera pandemia de H1N1 de 1917-1918, que causó unos 50 millones de muertes en todo el mundo.
COVID-19
El nombre dado a la enfermedad causada por el nuevo coronavirus (llamado SARS-CoV-2). Del inglés, COrona VIrus Disease 19 (debido al año de inicio de la pandemia, 2019). La COVID-19 se caracteriza por una alta transmisibilidad y un porcentaje de casos asintomáticos o leves. Tras un periodo de incubación de 2 a 14 días, pueden aparecer síntomas iniciales inespecíficos (tos, fiebre, pérdida de olfato, dolor de garganta, dolor de cabeza, fatiga y, finalmente, diarrea), que pueden ir seguidos de dificultad respiratoria, hipoxia e inflamación grave de las vías respiratorias, incluidos los pulmones.
Dado que se trata de una enfermedad todavía muy reciente, los conocimientos sobre el COVID-19 deben seguir estudiándose, especialmente en lo que respecta a los efectos a largo plazo, así como a las comorbilidades derivadas de la infección por el SARS-CoV-2, tanto en personas sintomáticas como asintomáticas. En casos más raros, puede producirse una encefalitis (inflamación del cerebro) y la afectación de otros tejidos, apareciendo en la literatura científica cuadros como la epididimitis y las complicaciones vasculares.
Evolución del genoma viral
Por evolución del genoma se entiende la suma de los procesos de: aparición de diferencias genéticas; acumulación de mutaciones; recombinaciones genéticas que pueden producirse cuando un mismo paciente es infectado por dos variantes de genética diferente; y, finalmente, el proceso de selección natural que actúa sobre las mutaciones genéticas favoreciendo aquellas que tienen algún tipo de ventaja sobre las demás, por ejemplo, una mayor capacidad infectiva.
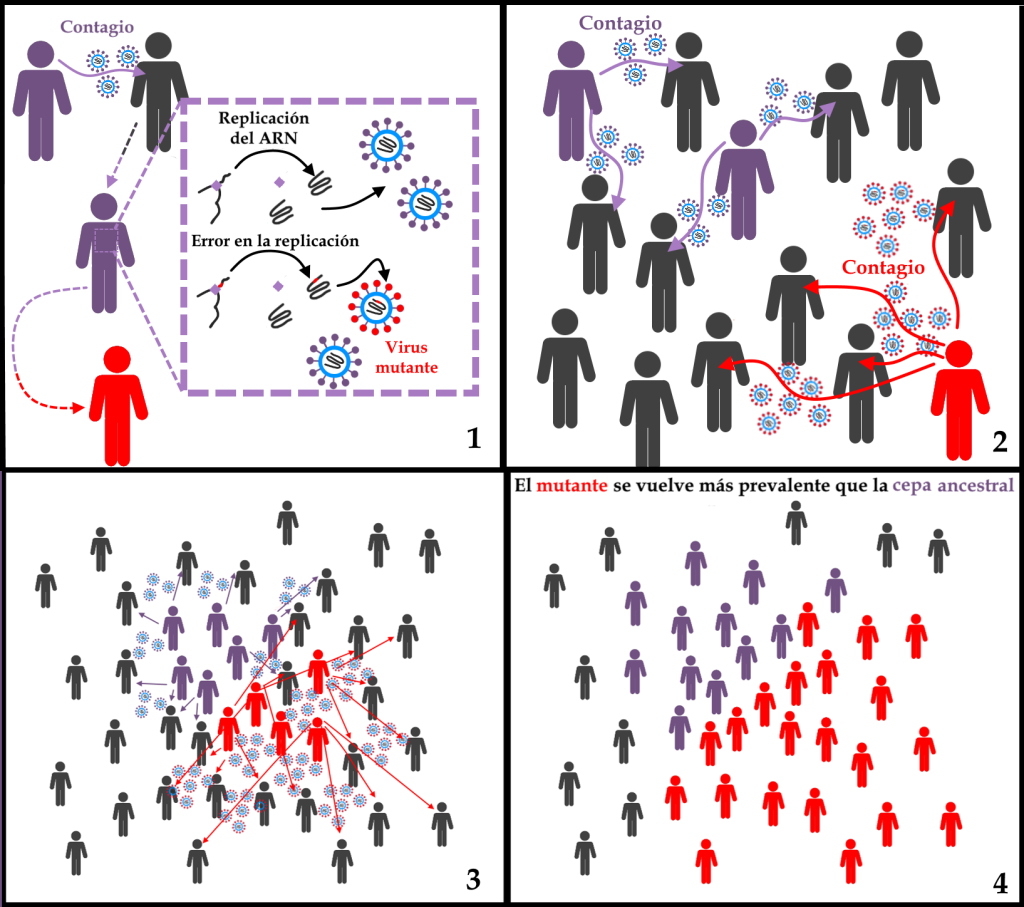
Diagrama que representa dos procesos de evolución viral. En el primer momento (1), debido a los errores aleatorios a los que siempre está sujeto el proceso de replicación del ARN, surge una muestra mutante. En el ejemplo hipotético del esquema, la alteración del genoma provoca un cambio en la proteína S, lo que proporciona una mayor infectividad del virus (2). Poco a poco, la ventaja que confiere la mayor infectividad (3) hace que el virus que contiene la mutación sea cada vez más común. Con cada nueva generación del virus, los mecanismos de selección natural siguen actuando, de modo que el mutante se vuelve más prevalente que su ancestro (4) debido a la ventaja que le confiere la mutación.
Seguir la evolución de un virus significa observar las nuevas cepas y cómo se diferencian unas de otras, qué proporción tiene cada una en la población total de la especie y cómo las tendencias epidémicas (crecimiento, estabilización o aumento del número de casos) se ven influidas por la distribución de las diferentes variantes y cepas de un virus.
La evolución de un virus no significa necesariamente que provoque un cuadro clínico más grave o que provoque una mayor mortalidad. En la mayoría de los casos, las mutaciones no tienen un impacto significativo en la biología del virus.
Filogenética
El estudio de las relaciones de parentesco evolutivo entre los organismos -incluidos los linajes virales- a partir del análisis de sus genomas se denomina “filogenética”. La filogenética es una forma de organizar las formas de vida y otras entidades biológicas en función de sus características y, antes del descubrimiento del material genético y del desarrollo de herramientas para estudiarlo, se realizaba comparando características de los organismos, como las estructuras de un mismo origen -por ejemplo, los brazos humanos y las aletas laterales de los delfines, que surgen de las mismas estructuras durante el desarrollo embrionario-. Más recientemente, las características que se comparan son generalmente genéticas, es decir, la estructura y funcionalidad de los genes en común entre diferentes entidades biológicas.
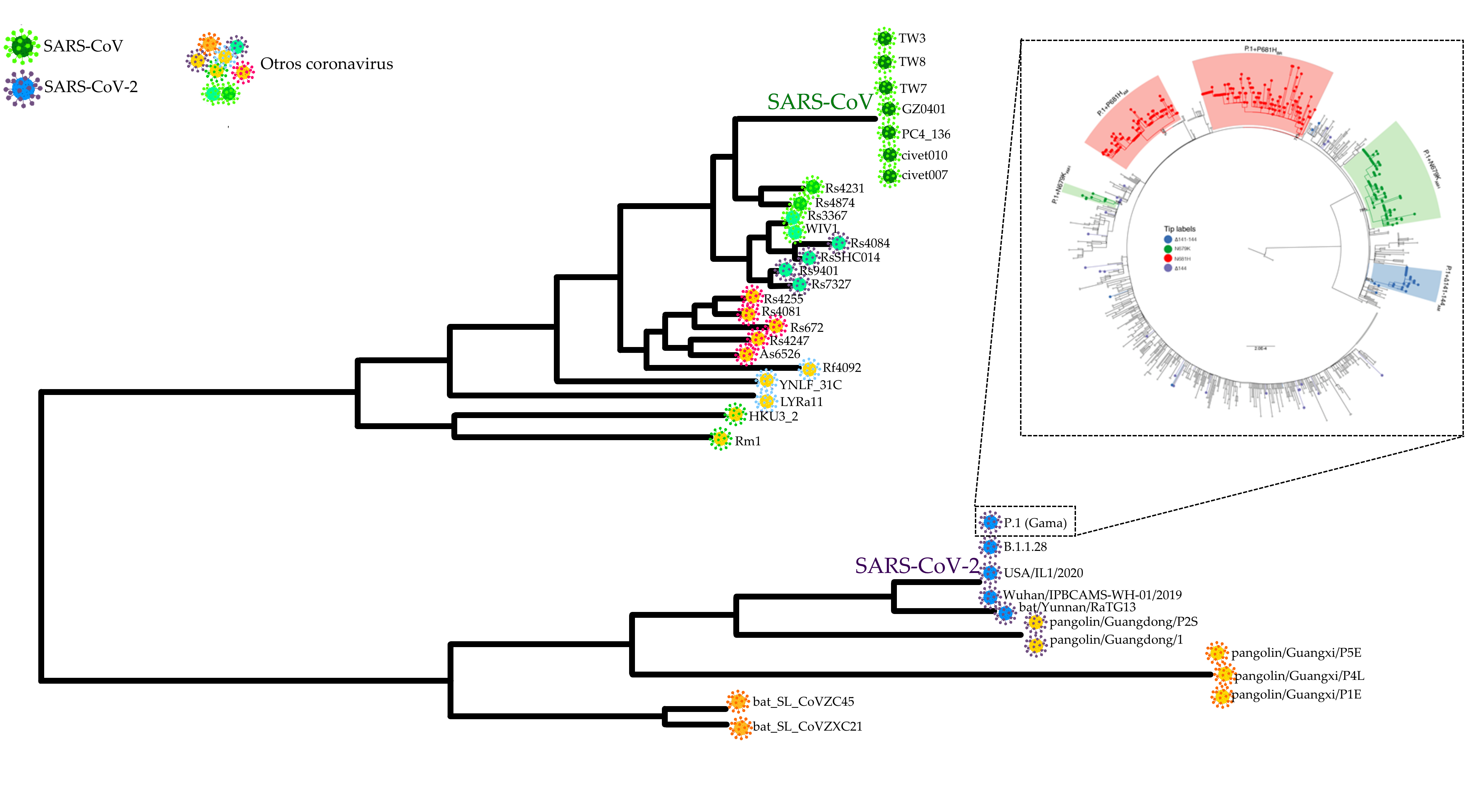 Esquema que representa las relaciones de parentesco entre diferentes muestras de coronavirus, incluidos los que causaron la epidemia de SARS en Asia entre 2002 y 2004 y la pandemia de COVID-19. El esquema más grande fue adaptado de Lauxmann et al. (2020), mientras que el esquema que detalla la evolución de los descendientes de la Variante Gamma (P.1) es una reproducción del esquema preparado por Naveca et al. (2021) para su publicación en el foro de expertos Virological.Org.
Esquema que representa las relaciones de parentesco entre diferentes muestras de coronavirus, incluidos los que causaron la epidemia de SARS en Asia entre 2002 y 2004 y la pandemia de COVID-19. El esquema más grande fue adaptado de Lauxmann et al. (2020), mientras que el esquema que detalla la evolución de los descendientes de la Variante Gamma (P.1) es una reproducción del esquema preparado por Naveca et al. (2021) para su publicación en el foro de expertos Virological.Org.
Inmunocomprometidos
Una persona que, por cualquier motivo, tiene una función reducida del sistema inmunitario. Entre las principales razones para comprometer la inmunidad se encuentran la desnutrición, los tratamientos médicos como el uso de corticosteroides o la quimioterapia, enfermedades como el VIH/SIDA, la leucemia y la aplasia medular, así como situaciones temporales como los efectos del estrés prolongado y la supresión de la inmunidad asociada naturalmente al embarazo.
Gene
Los genes son fragmentos del código genético que, al ser leídos y activados (o “expresados”), generan algún efecto en la célula. El efecto más clásico de un gen es la fabricación de una proteína por parte de una intrincada maquinaria celular, y la forma y función de esta proteína dependerá en gran medida de la estructura del gen que la originó. Los genes también pueden promover o silenciar la expresión de otros genes, así como, en el caso de los genomas basados en el ADN, dar lugar a moléculas de RNA que tendrán funciones efectivas dentro de la célula. Cuando, durante el proceso de duplicación del genoma, se produce un cambio en la estructura de un gen, este evento se denomina “mutación”.
Genética (campo de estudio)
La genética es el estudio de la herencia. De forma más directa, la genética estudia las estructuras del material genético de los seres vivos y de entidades biológicas como los virus, con la intención de entender cómo las secuencias dan lugar a sus características. El estudio de la genética puede centrarse en una variedad de escalas y objetivos, desde el estudio de un solo gen y la relevancia de su producto (el efecto cuando ese gen se expresa, o se “activa”) hasta el estudio de cómo las combinaciones de genes de una generación pueden influir en las características de su descendencia.
Genoma
A diferencia de la genética, el objetivo de la genómica es el estudio de los genes que interactúan entre sí para conformar una red integrada de secuencias que tienden a actuar de forma coordinada. En su totalidad, el genoma de un organismo o de un virus tiene múltiples genes. En los organismos con una genética más compleja, los genes pueden organizarse en grupos que se expresan o silencian juntos, así como en secuencias que regulan la intensidad de expresión de otros genes. Además de estudiar las secuencias de los genes individuales, el estudio del genoma implica comprender todas estas interacciones entre ellos, y cómo las alteraciones de este equilibrio, debidas a cambios en el entorno o a mutaciones en las propias secuencias, pueden afectar positiva o negativamente a los organismos.
Genoma de Alta Calidad (<1% de N)
Secuencia genómica resultante de una secuenciación precisa, en la que hay poca incertidumbre sobre los nucleótidos en cada posición del genoma. Cuando hay incertidumbre en la secuenciación, el software asociado al secuenciador registra “N” en la posición de la lectura del nucleótido ambiguo. Un porcentaje de N inferior al 1% significa que se conoce más del 99% de la secuencia genómica y es el resultado de una secuenciación fiable.
GISAID
GISAID (del inglés, Global Initiative for Sharing Data on Avian Influenza) es una iniciativa de cooperación científica internacional creada en 2008 para realizar esfuerzos más colaborativos y globales para vigilar y desarrollar estrategias para controlar los linajes circulantes de los virus de la Influenza (gripe).
Con una estructura de bases de datos y contactos entre instituciones de investigación dedicadas al estudio de la genómica viral, el GISAID amplió el alcance de sus actividades para incluir el nuevo coronavirus (SARS-CoV-2) y sus linajes en las actividades de seguimiento y cooperación. El objetivo de esta ampliación es permitir una lucha coordinada y más eficaz contra la propagación de la enfermedad mediante la comprensión de cómo se desarrolla la epidemia/pandemia, así como el seguimiento casi en tiempo real de los linajes del virus. La Red de Genómica de Fiocruz forma parte de la cooperación internacional de GISAID, y está generando y compartiendo datos sobre COVID-19 en Brasil.
Infección por contagio
Considerado por los autores de un artículo de revisión de 2017 como la característica definitiva de los patógenos que causan infecciones en los seres humanos a partir de otras especies de animales vertebrados (infecciones estas conocidas como “zoonosis”), el desbordamiento es el complejo proceso en el que un agente causante de una enfermedad en una especie o grupo biológico (como las aves, los mamíferos rumiantes, etc.) consigue adaptarse a otro grupo biológico de huéspedes. El proceso de propagación a otros huéspedes permite al patógeno causar infecciones/epidemias en esta nueva especie o grupo de especies, e incluso convertirse en endémico en sus poblaciones. El mismo artículo sostiene que, para que se produzca el spillover, es necesaria la alineación de varios factores, como la dinámica del patógeno en la población del huésped original, la dinámica de contacto entre los humanos y el huésped original (o los vectores de la enfermedad), las características del patógeno con respecto a su viabilidad fuera del huésped y las características individuales de la interacción del patógeno con el nuevo huésped (como la inmunidad y la compatibilidad molecular).
Letalidad
Número de muertes en la parte afectada de una población en un intervalo de tiempo determinado. Este indicador, a diferencia de la Mortalidad, solo tiene en cuenta la parte de la población afectada por la enfermedad (o el factor de riesgo), y no toda la población. Así, la Letalidad es una medida del riesgo de una causa para la totalidad de las personas afectadas -en el caso de COVID-19, por ejemplo, el indicador representa la posibilidad de que una persona infectada muera, mientras que la Mortalidad pone estas muertes en perspectiva en relación con toda la población.
Linajes (virus)
Conjunto de virus genéticamente relacionados que descienden de un ancestro común. Un linaje debe tener mutaciones que lo distingan de otras variantes del virus. Estas mutaciones no tienen por qué cambiar ninguna característica biológica del virus (por ejemplo, la transmisibilidad o el potencial de causar enfermedades). Para ser designada como cepa, debe tener relevancia epidemiológica, es decir, circular en una población amplia.
Material genético
En biología, la herencia -es decir, la transmisión de características de una generación a otra- se basa en el material genético. Es popular decir que algo está en el ADN, pero algunos virus, como los coronavirus y el VIH, por ejemplo, tienen su material genético basado en el RNA. Este tipo de molécula de ácido nucleico también está presente en nuestras células, pero interviene en los procesos del metabolismo celular y en el flujo de información genética, actuando, por ejemplo, en el proceso de producción de proteínas.
El material genético viral, cuando se inserta en la célula huésped, pronto es leído por la propia célula y da lugar a proteínas virales que inician el proceso de reorientación del metabolismo celular hacia la producción de nuevos virus. También se realizan nuevas copias del material genético viral a partir del material que entra en las células, y es debido a este proceso de realización de copias basado en el “molde” genético del virus que se pueden transmitir eventuales mutaciones.
Morbilidad
En epidemiología, el concepto de morbilidad difiere del que se utiliza normalmente en la práctica clínica, donde el término significa la gravedad de los síntomas de una enfermedad, o lo mucho que debilita a un paciente. En el contexto epidemiológico, la “morbilidad” denota la proporción de personas afectadas o portadoras de una enfermedad, en relación con la población general estudiada, en un lugar y momento determinados. Por ejemplo, si en un pueblo de 1000 habitantes, 100 personas están afectadas por una enfermedad, la morbilidad de esta enfermedad en la localidad es del 0,1 o del 10%.
Mortalidad
Número de muertes en una población en un intervalo de tiempo determinado. Cuando se trata de una causa concreta (como COVID-19), se habla de una Mortalidad específica de la causa. Es importante señalar que para este indicador se tiene en cuenta la población total (es decir, tanto los afectados por la causa de muerte en cuestión como los no afectados). De este modo, la mortalidad por causa específica ilustra el efecto de una causa de muerte sobre el total de la población estudiada, proporcionando una noción del riesgo sanitario colectivo. (Para una medida del efecto de una causa de muerte específicamente sobre la población afectada, véase letalidad)
Mutaciones
Las mutaciones son errores en el proceso de duplicación del material genético. Dado que el ADN (o RNA) que compone el genoma de una entidad biológica (ya sea un virus o un ser vivo) es “copiado” por enzimas que están sujetas a errores, pueden producirse pequeños cambios en el proceso, lo que da lugar a genomas ligeramente diferentes del original. La mutación es uno de los principales mecanismos de generación de diversidad en biología, ya que, si las copias fueran siempre exactas, no surgirían diferencias entre los organismos.
Estas diferencias suelen ser neutras o perjudiciales, pero si finalmente algún cambio aumenta el éxito de la siguiente generación, la tendencia es que esa mutación sea seleccionada y se haga más frecuente con el paso del tiempo. En el caso de un virus, por ejemplo, una mutación que da lugar a una mayor eficacia para entrar en las células del huésped, o en la estabilidad de la cápside viral -haciendo que sean viables durante más tiempo en el entorno celular- tiende a hacer que las variantes que la tienen sean más exitosas que las que no tienen la mutación (para un esquema que ilustra este proceso, véase Evolución del Genoma Viral, más arriba). Las mutaciones que provocan este tipo de diferencias suelen afectar directamente a la estructura de algún gen, la unidad fundamental de la información genética.
Una mutación también puede permitir que un virus entre en otros tipos de células, e incluso en otras especies, como en el caso del SARS-CoV-2, que probablemente se originó en los murciélagos y posteriormente desarrolló la capacidad de causar la enfermedad y transmitirse entre los humanos. Otros ejemplos son la gripe porcina y la gripe aviar, que tienen una historia similar.
Mutaciones que definen el linaje
Una mutación o un conjunto de mutaciones características de un linaje, verificadas por secuenciación genética en muestras pertenecientes a esa cepa. Las mutaciones que definen el linaje pueden dar lugar a genomas virales con características distintas, como una mayor o menor transmisibilidad, según los genes afectados.
Pandemia
Aunque el término “Pandemia” tiene un significado lato sensu, una revisión de 2009 destinada a comprender los atributos clave que hacen que un evento epidémico se considere realmente una “Pandemia” enumera las siguientes características:
- “Amplia difusión geográfica” (evento sin fronteras);
- “Movilidad y expansibilidad de la enfermedad” (es decir, la capacidad de ser transmitida competentemente por los huéspedes de una localidad a otra);
- “Altas tasas de ataque y transmisión en brotes/irrupciones” (aparición de múltiples casos en un intervalo de tiempo corto);
- “Inmunidad reducida (o ausente) en la población”;
- “Originalidad y evasión del agente etiológico” (no necesariamente aparición de una nueva especie, puede tratarse de una evolución, de un nuevo linaje o serotipo, o simplemente del recrudecimiento del mismo patógeno después de un cierto periodo de tiempo en el que la población ya no es inmunológicamente competente);
- “Infectividad” (la capacidad de causar una infección y establecerse);
- “Contagio” (capacidad de transmitirse directamente de una persona a otra, con la salvedad de que pandemias como la peste bubónica durante la Edad Media tenían la vía vectorial como principal vía de transmisión);
- “Gravedad de los síntomas y letalidad” (con alto impacto en la capacidad productiva de la población y tasas de mortalidad significativas);
Es más que plausible atribuir todas las características anteriores de una “Pandemia” a ésta causada por la aparición del nuevo coronavirus humano, llamado SARS-CoV-2 y causante de la Enfermedad por Coronavirus 2019, simplemente llamada COVID-19. La COVID-19 es una lesión muy preocupante, que a menudo provoca cuadros inflamatorios graves y la muerte. Su probable origen es zoonótico, y su alta adaptabilidad para infectar al huésped humano, al ser de transmisión aérea, combinada con el hecho de que es una nueva especie de virus (originalidad) a la que nunca ha habido una exposición previa y, en consecuencia, ninguna inmunidad adquirida (población inmunológicamente virgen – también llamada por el término técnico naïve), contribuyó a su rápida propagación por todos los continentes, facilitada también por la amplia movilidad de las poblaciones anfitrionas debido a una economía globalizada y a un bajo nivel de preparación y respuesta (preparedness & response) de los sistemas de vigilancia de la mayoría de los países, que no tenían forma de anticipar el nuevo patógeno, ni pudieron actuar con la suficiente rapidez.
En términos técnicos, si posteriormente un agente causante de una Pandemia (o Epidemia) se establece localmente en una población determinada en aparente perennidad, provocando eventualmente ciclos de infección, ya sea estacional o esporádica, sin erradicación total del agente etiológico, este patrón se denomina “Endémico”.
Partículas de virus infecciosas
Partículas virales capaces de multiplicarse en una célula y/o tejido, generando un proceso infeccioso, con la estructura completa, incluyendo las proteínas de la cápside, la envoltura fosfolipídica (cuando está presente), las proteínas de superficie o glicoproteínas -que intervienen en el proceso de invasión de las células huésped- y el genoma viral completo.
En teoría, una sola partícula vírica viable es capaz de iniciar el proceso de infección cuando encuentra una célula huésped compatible, aunque lo más habitual es que sean necesarias varias partículas que entren en el organismo de un huésped para que el proceso de infección tenga éxito.
Partículas no viables
Partículas virales que, debido a la ausencia de uno o más de sus constituyentes, son incapaces de completar el ciclo viral con éxito. Esta incapacidad puede deberse, por ejemplo, a la producción de cápsides vacías (sin material genético), o a la ausencia de una glicoproteína de superficie importante para el proceso de invasión de las células huésped, o a moléculas defectuosas (ya sea con material genético incompleto o inviable, o con proteínas o glicoproteínas cuya función está comprometida por mutaciones deletéreas).
Patógeno endémico / Infección endémica
Una enfermedad (o el agente patógeno causante de la misma) puede considerarse endémica en una localidad cuando está presente de forma regular en ella, con una baja prevalencia. Eventualmente, una enfermedad endémica puede causar brotes en la región -es decir, tener un aumento en el número de casos-, pero a diferencia de los brotes de enfermedades no endémicas, tras un descenso en el número de casos la enfermedad endémica sigue circulando entre
reservorios y anfitriones y se mantiene en la baja prevalencia característica de una endemia.
Preimpresión
Una publicación inicial que contiene el texto y los datos similares a un artículo terminado, pero que se publica antes del proceso de revisión por pares. Al no haber sido sometido al riguroso proceso de verificación y revisión independiente por parte de otros grupos de investigación -que forma parte del proceso editorial habitual para la publicación de artículos científicos-, las conclusiones de un preimpreso son limitadas.
Este modelo de publicación es importante para compartir rápidamente la información entre los expertos en medio de una crisis como la actual pandemia, y puede informar a los medios de comunicación y a la opinión pública siempre que la información sea tratada con mayor cautela y responsabilidad, destacando el hecho de que todavía es necesario el proceso editorial completo para obtener más certeza en las conclusiones presentadas.
Prevalencia
La prevalencia de una enfermedad o de un linaje viral es una medida del número de personas afectadas por la enfermedad en un periodo de tiempo determinado. Un linaje es más prevalente que otro cuando, en un momento dado, hay más personas enfermas por ella que por la otra.
Profundidad de lectura
Una medida de la fiabilidad de una secuencia genética. La profundidad de lectura es el número de veces que se ha detectado un nucleótido en esa posición entre todas las lecturas de un proceso de secuenciación. Una profundidad elevada significa que la probabilidad de error (es decir, que un nucleótido en una posición determinada no sea el objetivo del secuenciador) es baja.
R0
También llamado Número Básico de Reproducción, en epidemiología R0 se define como la capacidad de propagación de un agente etiológico, asumiendo que la población es el 100% susceptible a la enfermedad. La cifra es una estimación de cuántas personas, por término medio, se infectan al entrar en contacto con un paciente infectado. Por lo tanto, un R0 de 1 significa que cada persona, durante el periodo de infección, es capaz de transmitir la enfermedad a una sola persona.
Cuando el R0 es superior a 1, la tendencia es que la enfermedad se extienda hasta provocar un brote (el número de casos aumenta a medida que cada paciente infecta a más de una persona), mientras que un R0 inferior a 1 significa una tendencia a que la enfermedad desaparezca.
Re
ientras que R0 es una medida de la máxima propagación potencial de una enfermedad infecciosa, suponiendo que no hay inmunidad en la población ni medidas para contener el contagio, Re es una estimación del número real de contagios. La incidencia puede variar a lo largo del tiempo, según el desarrollo de la inmunidad (por vacunación o en respuesta a la infección), y la adopción de políticas públicas de contención y profilaxis de la enfermedad, medidas que conducen a la prevención de la infección.
En el contexto de Covid-19, las medidas preventivas, como el distanciamiento social con la adopción de trabajos no presenciales, la interrupción temporal de las actividades escolares y, en situaciones más extremas, el encierro (situación en la que se suspenden todas las actividades presenciales no esenciales y se restringe el movimiento de la población a situaciones de extrema necesidad), tienen como objetivo reducir el Re, es decir, reducir el número de personas efectivamente contaminadas por el contacto con un paciente.

Cuando se trata de enfermedades infecciosas como el COVID-19, los eventos en los que se reúnen muchas personas pueden resultar en un aumento de Re, debido al gran contacto entre las personas infectadas y otras personas susceptibles. Eventos como este se denominan “Eventos supercontagiadores”.
Reservorio
En epidemiología, se dice que un animal, una persona o incluso un factor ambiental (por ejemplo, una masa de agua contaminada) es un reservorio de una enfermedad cuando mantiene en circulación a largo plazo el agente causante de la misma. El agente infeccioso puede transmitirse, directa o indirectamente, de los reservorios a los huéspedes de la enfermedad, lo que puede provocar brotes e incluso pandemias. Cuando se trata de un reservorio biológico (persona o animal), no es raro que el propio reservorio no enferme, sino que solo sirva de “puente” para la transmisión de la enfermedad a los huéspedes susceptibles. Los reservorios también son un factor importante que dificulta la erradicación de una enfermedad, ya que, aunque el patógeno se elimine por completo entre los huéspedes de la enfermedad, seguirá circulando entre los reservorios.
RT-PCR
Examen utilizado para detectar la presencia y la cantidad de ARN en muestras biológicas. Mediante el uso de moléculas de ARN sintetizadas en el laboratorio, denominadas “sondas” y “cebadores”, que se unen específicamente a una secuencia genética de interés -en este caso, tramos del genoma del SARS-CoV-2- es posible detectar si hay ARN viral en la muestra y cuantificar el número de copias de la secuencia de interés mediante el uso de la fluorescencia.
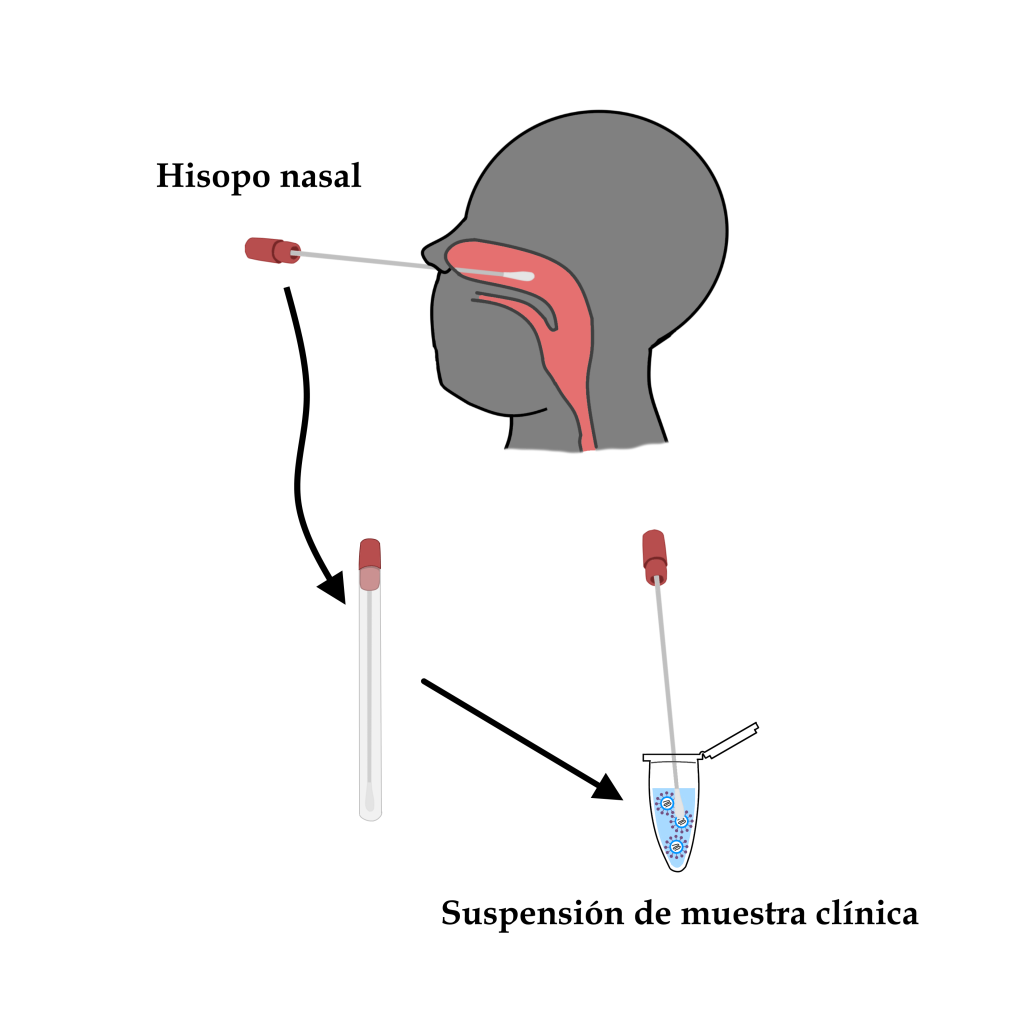
La obtención de una muestra clínica para la detección de material genético viral se realiza mediante la técnica del hisopo nasal. Con un hisopo adecuado, los profesionales sanitarios recogen material de la mucosa de la orofaringe del paciente en un caso sospechoso, del que se obtiene una suspensión de partículas virales.
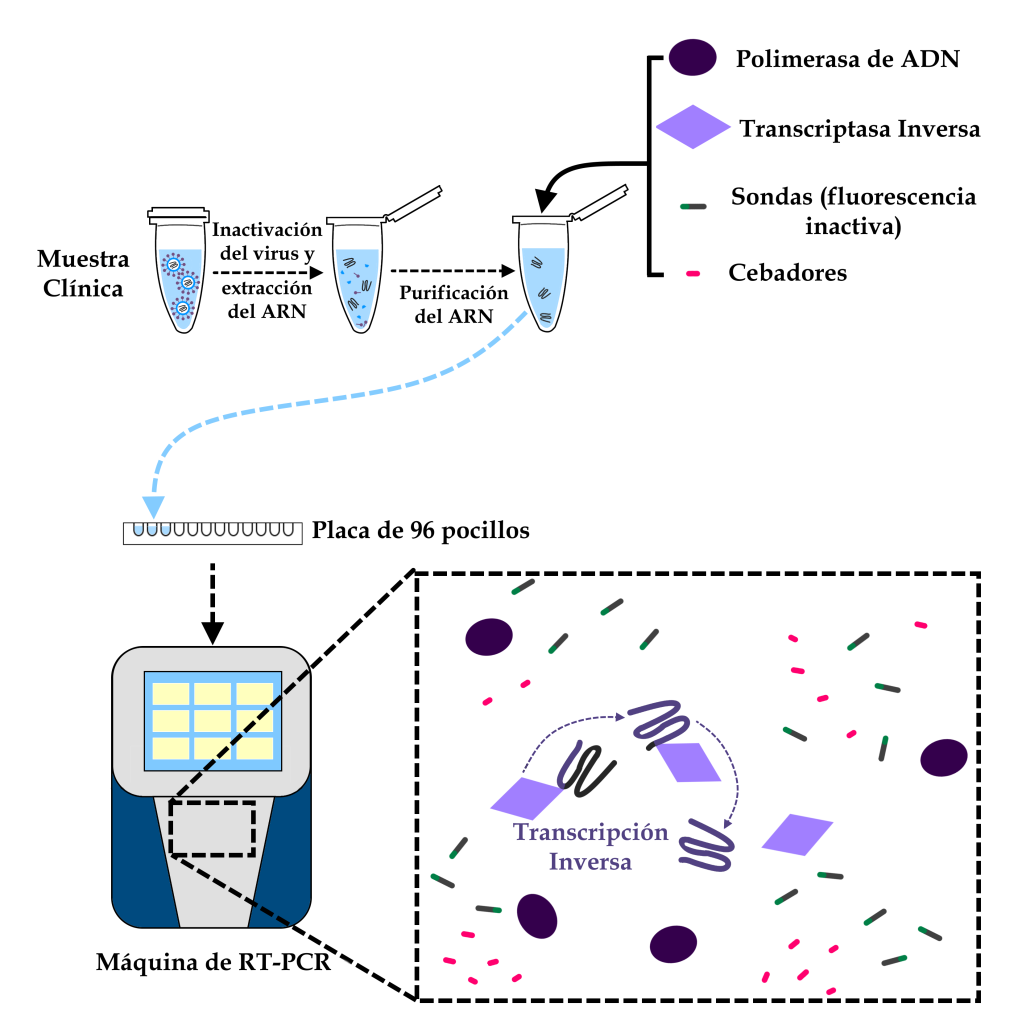
A partir de la muestra clínica, es posible realizar la extracción y purificación del ARN viral. Con el ARN purificado, se añaden a la mezcla los reactivos necesarios para realizar la RT-PCR, que se distribuyen en una placa acrílica específica para este uso. En el equipo de RT-PCR, el llamado ADN complementario se obtiene mediante la reacción de la transcriptasa inversa (RT, que da nombre a la técnica). Es este ADN complementario -una copia fidedigna del material genético viral- el que servirá de base para los siguientes pasos.
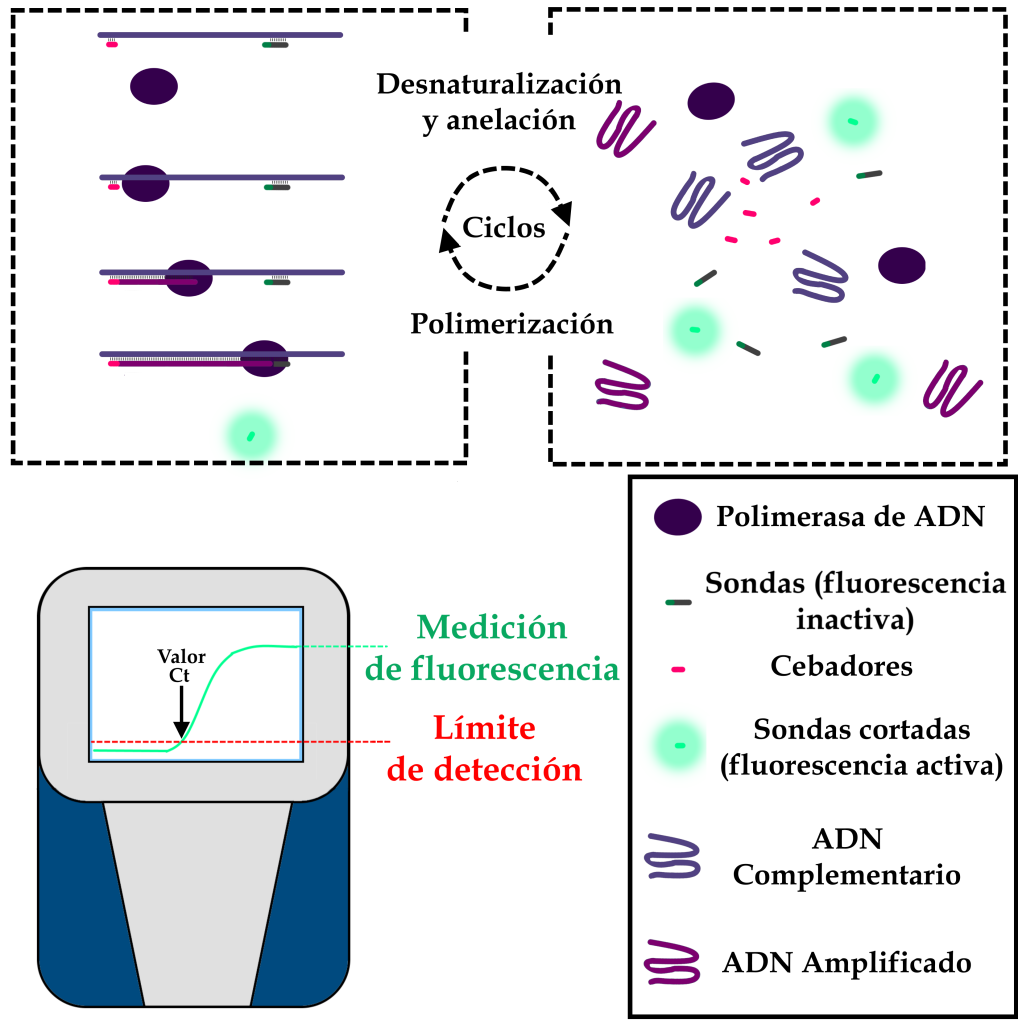
El aparato de RT-PCR realiza varios ciclos, durante los cuales los descensos y aumentos controlados de la temperatura de las muestras permiten que las enzimas de la ADN-polimerasa hagan copias del ADN complementario. Al copiar las secuencias, la enzima promueve la rotura de las sondas, lo que activa los marcadores fluorescentes. De este modo, la intensidad de la fluorescencia en cada ciclo es directamente proporcional a la cantidad de material genético amplificado. Cuando la intensidad de esta fluorescencia es suficiente para que los sensores del aparato la detecten, se dice que se ha alcanzado el umbral de fluorescencia. El número de ciclos necesarios para que una muestra alcance este umbral se denomina “valor Ct”.
SARS-CoV-2
El CoronaVirus del Síndrome Respiratorio Agudo Severo 2 (en inglés, Severe Acute Respiratory Syndrome CoronaVirus 2) es el segundo coronavirus responsable de una epidemia de síndrome respiratorio en humanos, tras el brote de 2003 en China causado por el SARS-CoV. El síndrome respiratorio agudo severo es un cuadro clínico que supone un alto riesgo para la salud y puede provocar la muerte.
Todos los linajes y variantes del nuevo coronavirus pertenecen a la especie SARS-CoV-2.
Semana epidemiológica
El calendario de semanas epidemiológicas es un sistema de normalización del recuento de semanas, para facilitar la comparación entre diferentes años en lo que respecta a las estadísticas epidemiológicas (por ejemplo, comparar la mortalidad de una enfermedad entre dos o más años). Por convención internacional, las semanas epidemiológicas se cuentan de domingo a sábado, siendo la primera semana del año la que contiene más días en enero y la última la que contiene más días en diciembre.
Secuenciadores
Equipo que permite desvelar la secuencia de partes o la totalidad de un genoma mediante la lectura de las secuencias de nucleótidos (“bloques” a partir de los cuales se construye el ADN o el RNA). Los secuenciadores se basan en la reacción de síntesis del código genético, que se produce de forma muy similar cuando las células se dividen, combinada con tecnologías para identificar qué nucleótido se inserta cada vez. La obtención de secuencias de tramos del genoma, o de todo su contenido, permite comparar diferentes muestras, entender cómo funcionan las relaciones entre los genes y clasificar las muestras según su parentesco.
Secuencias de consenso
También conocidas como secuencias canónicas, las secuencias de consenso, en biología molecular, son el resultado de comparar varias secuencias genómicas alineadas. Para formar una secuencia de consenso, se consideran los nucleótidos más frecuentes en cada posición. Así, las secuencias de consenso pueden servir de referencia para el análisis de nuevas muestras.
Sistema de nomenclatura
Conjunto de reglas que deben seguirse para nombrar una variante recién encontrada de un virus. Para el SARS-CoV-2, la Red Genómica de Fiocruz sigue el sistema de nomenclatura PANGO propuesto por el Centro de Vigilancia Genómica de Patógenos (Inglaterra).
Seroconversión
También conocida como “seroconversión inmunitaria”, se produce cuando los anticuerpos específicos -producidos en respuesta a la exposición natural o a la vacunación contra un agente infeccioso o una toxina- pueden detectarse en el plasma sanguíneo de un paciente. En general, la producción de anticuerpos, fabricados por las células denominadas linfocitos B, comienza tras un intervalo de tiempo de unos días tras la exposición. En las primeras etapas, lo más habitual es que se produzcan anticuerpos de tipo IgM, y más tarde, anticuerpos de tipo IgG, de modo que es posible distinguir entre los eventos de seroconversión recientes (presencia de IgM y poca o ninguna IgG) y los eventos más antiguos (caracterizados por la presencia de mayores cantidades de IgG, y de IgM en pequeñas concentraciones, o incluso indetectables).
SNP
El polimorfismo de un solo nucleótido es la sustitución de un solo nucleótido en un gen al comparar dos o más muestras. Si, por ejemplo, en una población, en la posición X de un gen puede haber una timina (T) o una adenina (A), se puede decir que hay un SNP en esta posición del gen. Estas diferencias pueden influir en el éxito evolutivo de los virus a través de: cambios en las proteínas que confieren ventajas o desventajas adaptativas -por ejemplo, los diferentes cambios en la proteína S que permiten a algunas variantes del SARS-CoV-2 causar reinfección-; o un escape a los mecanismos de defensa de las células del huésped contra el material genético invasor (como los ARN de interferencia).
Tasa de transmisión
En los modelos epidemiológicos, la tasa de transmisión es la proporción de individuos de una población clasificados como “susceptibles” que pasan al estado “infectado”. La tasa de transmisión, por lo tanto, representa la propagación de una epidemia a través de la población, y es diferente de R0 y Re, aunque está directamente relacionada con ambos – por ejemplo, las medidas de distanciamiento y la vacunación, al reducir los valores de Re, también reducen la tasa de transmisión. Otro ejemplo es que los patógenos con un R0 más alto conducen a tasas de transmisión más altas, dado que las demás condiciones (como la intensidad y el cumplimiento de las medidas de aislamiento y la susceptibilidad de la población) son las mismas.
Valor Ct
En una reacción de PCR (ya sea cuantitativa en tiempo real o convencional), el equipo realiza ciclos de cambio de temperatura porque cada paso de la amplificación del material genético se optimiza a una temperatura diferente. En una RT-PCR, es posible hacer un seguimiento de cuántos ciclos pasan hasta que hay suficiente material genético para ser detectado por el dispositivo. Como una mayor cantidad de RNA en una muestra significa que se necesitan menos ciclos para alcanzar este límite mínimo de detección, el número de ciclos, que se denomina valor Ct (umbral de ciclo).
De este modo, dado que el valor Ct es inversamente proporcional a la cantidad de material genético analizado, en el ámbito de la monitorización del SARS-CoV-2, se pueden comparar diferentes muestras en cuanto a su valor Ct para evaluar cuáles tienen una mayor carga viral (menor valor Ct). Es importante tener en cuenta que no se trata de una medida directa del número de copias del virus en el cuerpo de los pacientes, sino de un indicador consistente que permite el análisis comparativo.
Variantes genéticas
Cualquier virus que haya sido secuenciado y tenga mutaciones que lo diferencien de la versión original del virus. El término variante no implica importancia epidemiológica y puede utilizarse en sentido amplio para referirse a la diversidad genética de una especie viral, es decir, al repertorio de diferentes versiones de sus genes. Los medios de comunicación también utilizan el término cepa para designar las variantes víricas, aunque el uso de este término no está consensuado en la comunidad virológica.
Algunas de las variantes y linajes más relevantes para el escenario brasileño son:
B.1.1.28
Uno de los linajes del SARS-CoV-2 con probable origen en Brasil, el B.1.1.28 se detectó por primera vez el 5 de marzo de 2020. De este linaje se derivó el VOC Gamma (P.1), que ha causado preocupación debido a su capacidad de causar reinfecciones en algunos pacientes, así como cargas virales más altas.
B.1.1.33
El linaje B.1.1.33, la principal que circula en Brasil durante 2020. Tiene su origen en casos asociados a una variante ancestral -probablemente de Europa- llamada B.1.1.33-like. Este linaje ancestral ya tenía una de las dos mutaciones que definen el linaje B.1.1.33 (T29148C/I292T, en la proteína de la nucleocápside), y se cree que la segunda de estas mutaciones apareció en cuestión de semanas después de la aparición de los primeros casos en Brasil en febrero de 2020 (T27299C/I33T, en ORF6). El linaje B.1.1.33 alcanzó una prevalencia superior al 80% en el estado de Río de Janeiro, mientras que en estados como Ceará y Pernambuco su prevalencia fue menor: 3% y 2%, respectivamente.
B.1.195
Linaje inicialmente dominante en el estado de Amazonas, pero posteriormente suplantado por B.1.1.28 (que luego fue suplantado por la variante Gama)
Variante Gama / P.1
Variante del linaje B.1.1.28 detectada inicialmente en enero de 2021, en muestras de turistas japoneses que acababan de regresar a casa después de visitar el estado de Amazonas. El origen más probable de la variante es en el propio Amazonas, hacia principios de diciembre de 2020. La rápida propagación a varias regiones de Brasil y, posteriormente, a otros países, junto con las mutaciones de la cepa, ha hecho que Gama sea clasificada como Variante Preocupante (VOC). Entre las 21 mutaciones que definen el linaje de la variante, los cambios en el dominio de unión al receptor (RBD, del inglés Receptor Binding Domain) y en la glicoproteína Spike (proteína S) dan lugar a una proteína lo suficientemente diferente como para reducir la capacidad de los anticuerpos adquiridos en infecciones anteriores o en la vacunación para neutralizar la infección, lo que puede contribuir a la aparición de reinfecciones. Gamma tiene 12 mutaciones en la proteína S, que incluyen tres mutaciones preocupantes en común con el linaje Beta (B.1.351): a saber, K417N / T, E484K y N501Y. Los datos preliminares apuntan además a una posible mayor carga viral en los pacientes infectados con la variante Gamma. Si se confirma este hallazgo, es posible que la mayor transmisibilidad de esta variante esté relacionada con la interacción de varios factores: el aclaramiento de un mayor número de partículas víricas, una posible mayor afinidad de la proteína S por el receptor presente en las células humanas y la circulación de Gamma incluso entre la población recuperada de anteriores infecciones por SARS-CoV-2.
Variante P.2
Variante del linaje B.1.1.28 detectado por primera vez en Río de Janeiro, inicialmente clasificada como Variante de Interés (VOI) bajo el nombre de “Zeta”, pero actualmente sin clasificar como VOC, VOI o VUM. La P.2 tiene 5 mutaciones que definen el linaje, incluida la sustitución de aminoácidos S:E484K, presente en variantes como la B.1.1.7+E484K británica y la Gamma (P.1), y aparentemente asociada a la capacidad de causar reinfección debido a diferencias en la estructura de la proteína S que reducen la neutralización por los anticuerpos. Las inferencias derivadas del estudio de la filogenia de los linajes que circulan en Brasil trazan la aparición de P.2 aproximadamente en julio de 2020. En octubre del mismo año, la variante se vuelve expresiva entre las muestras recogidas en el estado de Río de Janeiro, llegando a ser dominante en el estado en diciembre de 2020. A partir de febrero de 2021, el predominio epidemiológico de la variante P.2 es suplantado por la llegada de Gama al estado, que tuvo un rápido aumento en el número de casos.
Variante P.3
Variante inicialmente clasificada como Variante de Interés (VOI) denominada “Theta” e identificada por primera vez en Filipinas; P.3 tiene algunas mutaciones también presentes en las variantes de interés, como E484K y N501Y, y otras 5 mutaciones definidoras de linaje de un total de 7. Actualmente ya no tiene la clasificación de riesgo aumentado y no es ni VOC ni VOI ni VUM.
Variante P.4
Variante aún no clasificada como VOC o VOI, descubierta en mayo de 2021 en el interior de São Paulo en un estudio de caracterización genómica de muestras de SARS-CoV-2 realizado por investigadores de la Universidad Estadual Paulista. Genéticamente cercana a otras variantes “P”, como Gama (P.1), P.2 y P.3, P.4 tiene entre sus mutaciones la sustitución L452R, asociada a una mayor infectividad y a un potencial escape de anticuerpos.
Variante Delta / B.1.617.2
La variante clasificada como VOC, Delta, se detectó inicialmente en el subcontinente indio, pero al igual que otras variantes del SARS-CoV-2, pronto se extendió a otros países debido al tráfico de personas. La variante Delta tiene las mutaciones T19R, (G142D*), 156del, 157del, R158G, L452R, T478K, D614G, P681R, D950N en la proteína S, siendo L452R y P681R mutaciones de relevancia biológica. La suma de las diferencias en VOC Delta hace que esta variante sea más transmisible, y posiblemente escape a la respuesta de los anticuerpos, que otras VOC. Como el proceso evolutivo sigue ocurriendo en las variantes, con la selección de muestras portadoras de mutaciones ventajosas, en algunas localidades circula con mayor intensidad una variación de Delta, llamada popularmente “Delta plus”, portadora de la mutación de interés K417N, también observada en las variantes Beta y Gamma. Tanto Delta como sus descendientes están asociados al empeoramiento del número de casos y muertes en países como India y Estados Unidos, por lo que la llegada de esta variante a Brasil puede suponer un riesgo para la salud pública, teniendo en cuenta que la cobertura de vacunación completa (porcentaje de la población que ha recibido una inmunización completa) es todavía muy baja en el país.
Variante B.1.617.1
La variante inicialmente clasificada en su momento como VOI, bajo el nombre de Kappa, B.1.617.1 se detectó inicialmente en el subcontinente indio, pero, al igual que otras variantes del SARS-CoV-2, pronto se extendió a otros países debido al tráfico de personas. La variante B.1.617.1 tiene las mutaciones (T95I), G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H en la proteína S. Estos cambios pueden conducir a la evasión de la respuesta de los anticuerpos, pero esta capacidad aún se está estudiando. Actualmente, la OMS ya no clasifica esta variante como VOI o VUM.
Mu / B.1.621 variante
Variante hasta ahora clasificada como VOI. Mu se detectó inicialmente en muestras de Colombia, pero, al igual que otras variantes del nuevo coronavirus, ha avanzado a otros países, más expresivamente en América Latina (por ejemplo, Ecuador, 13% de las muestras analizadas hasta septiembre de 2021; Chile, ~40% de las muestras desde agosto de 2021). La variante tiene en su genoma mutaciones asociadas a muestras dotadas de capacidad de neutralización por anticuerpos, como E484K y K417T (también presente en la variante Gamma), además de otras alteraciones genéticas aún en estudio. La variante Mu ha llegado a Brasil, aunque todavía no se ha detectado un volumen tan expresivo como el de otras variantes como Gamma y Delta.
Variante Ómicron / B.1.1.529
Variante preocupante (VOC) detectada por primera vez en Sudáfrica y Botsuana en noviembre de 2021. El análisis del genoma de las muestras de VOC Ómicron revela el mayor número de mutaciones hasta la fecha entre las variantes de interés, con unas 30 solo en la proteína Spike. Una de estas mutaciones es una deleción en la región 69/70 de Spike, asociada al fracaso de la amplificación por PCR de este gen. Esta característica se ha utilizado para inferir la frecuencia y controlar la propagación de Ómicron, de forma similar a como se hizo con VOC Alpha.
Se ha demostrado que VOC Ómicron tiene una transmisibilidad muy alta y se ha asociado a una nueva ola de COVID-19 en Sudáfrica, donde ha superado a VOC Delta, y a un rápido aumento de los casos en Inglaterra. La OMS comenta que hay motivos de preocupación por la presencia de mutaciones como las de otros VOC, que se asocian a una nueva fuga de la inmunidad generada por infecciones anteriores con el SARS-CoV-2 o por la acción de los inmunizadores (vacunación), pero subraya que la vacunación y la prevención de la infección son las formas más eficaces de evitar los casos graves y las hospitalizaciones.
Virus
Grupo de entidades biológicas no celulares. Al no poseer un metabolismo propio (es decir, sin orgánulos ni enzimas que lleven a cabo reacciones químicas, generen o consuman energía), los virus son parásitos, completamente dependientes de las células del huésped para su replicación, el proceso de creación de nuevas copias, análogo a la reproducción. Así, todo virus está formado por material genético rodeado de una estructura proteica llamada cápside.
Algunos virus -como el que causa el COVID-19, el SARS-CoV-2- tienen una capa de fosfolípidos llamada “envoltura”. La envoltura es similar a la de la célula en el sentido de que partes de la membrana de la célula huésped son cooptadas por estos virus con envoltura para formar esta estructura al final del proceso de replicación. Con o sin envoltura, todo virus necesita tener en su superficie moléculas -generalmente proteínas o glicoproteínas- que le sirvan para “aferrarse” a las células del huésped y colocar en su interior el material genético viral, que puede estar basado en ADN o RNA, y que contiene toda la información necesaria para causar la infección (incluidos los genes para la producción de las enzimas y proteínas virales, a través de los cuales los parásitos dominan el metabolismo de las células del huésped y lo redirigen a la producción de nuevos virus).
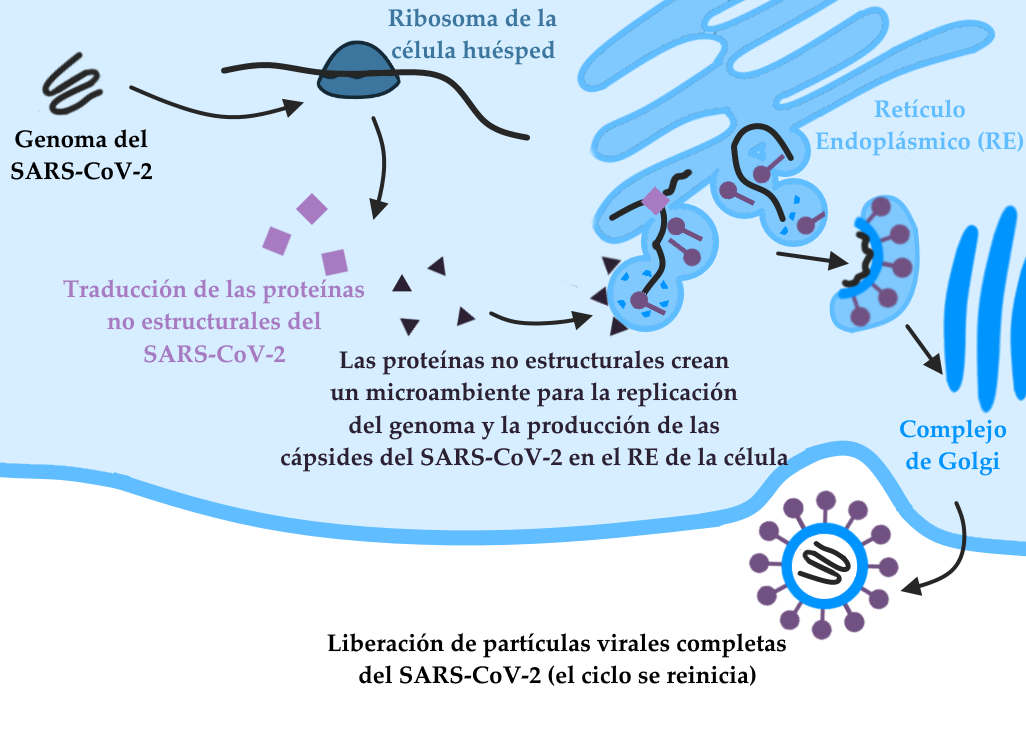
Esquema que ilustra las etapas de la infección tras la entrada del genoma viral en la célula huésped. Obsérvese que la envoltura viral es el resultado de una mezcla de proteínas del virus y lípidos de la membrana de la célula huésped.
VOC
Las Variantes Preocupantes (del inglés, Variants of Concern) es una clasificación que se da a las variantes de una cepa cuando se demuestra que tiene una mayor transmisibilidad (o efectos negativos en la epidemiología del virus), virulencia (incluyendo cambios en la presentación clínica de la enfermedad) o una menor eficacia de las medidas de salud pública (incluyendo el distanciamiento social y las opciones diagnósticas, vacunales y terapéuticas actualmente disponibles) contra la variante en cuestión. Así, variantes como la Gamma (P.1), la Alfa (B.1.1.7) y la Beta (B.1.351), debido a su rápida difusión en sus países de origen, al elevado número de mutaciones en la proteína S y al creciente número de evidencias de su capacidad de escape de anticuerpos, fueron clasificadas como VOC.
VOI
Las Variantes de Interés (VOI, del inglés Variants of Interest) son una clasificación que se da a los perfiles de genes mutantes con el potencial de generar un empeoramiento del cuadro epidémico, pero que aún no se ha demostrado que tengan este potencial. La línea de corte de la clasificación es baja -es decir, bastan unas pocas mutaciones en genes relevantes para que una muestra reciba esta clasificación- para que no pasen desapercibidas las variantes con posibilidad de causar un empeoramiento del problema de salud. Para ser clasificada como VOI, una variante tiene que tener una mutación que genere cambios de aminoácidos posiblemente asociados a cambios en la transmisibilidad, virulencia (capacidad de generar síntomas graves), epidemiología o en la capacidad de ser reconocida por el sistema inmunitario, además de haber causado múltiples casos de la enfermedad/transmisión comunitaria.
VUM
Las Variantes bajo Vigilancia (VUM, del inglés Variants under Monitoring) es una clasificación que se da a los perfiles genéticos mutantes del SARS-CoV-2 que no presentan necesariamente riesgo, pero que tienen características que justifican una vigilancia más cautelosa. La Organización Mundial de la Salud clasifica las cepas como VUM cuando se sospecha que algunas de sus alteraciones genéticas tienen efectos sobre las características virales potencialmente arriesgadas en el futuro, pero sin pruebas concluyentes sobre el mayor riesgo asociado a estas mutaciones según los conocimientos actuales.